Sample QC Sub-workflow¶
- Workflow File:
Config Options: see The config.yml for more details
reference_files.thousand_genome_vcf
reference_files.thousand_genome_tbi
software_params.sample_call_rate_1
software_params.snp_call_rate_1
software_params.sample_call_rate_2
software_params.snp_call_rate_2
software_params.intensity_threshold
software_params.contam_threshold
software_params.ibd_pi_hat_min
software_params.ibd_pi_hat_max
software_params.maf_for_ibd
software_params.ld_prune_r2
software_params.dup_concordance_cutoff
software_params.pi_hat_threshold
workflow_params.remove_contam
workflow_params.remove_rep_discordant
Major Outputs:
sample_level/sample_qc.csv
sample_level/snp_qc.csv
sample_level/concordance/KnownReplicates.csv
sample_level/concordance/InternalQcKnown.csv
sample_level/concordance/StudySampleKnown.csv
sample_level/concordance/UnknownReplicates.csv
sample_level/summary_stats.txt
sample_level/call_rate.png
sample_level/chrx_inbreeding.png
sample_level/ancestry.png
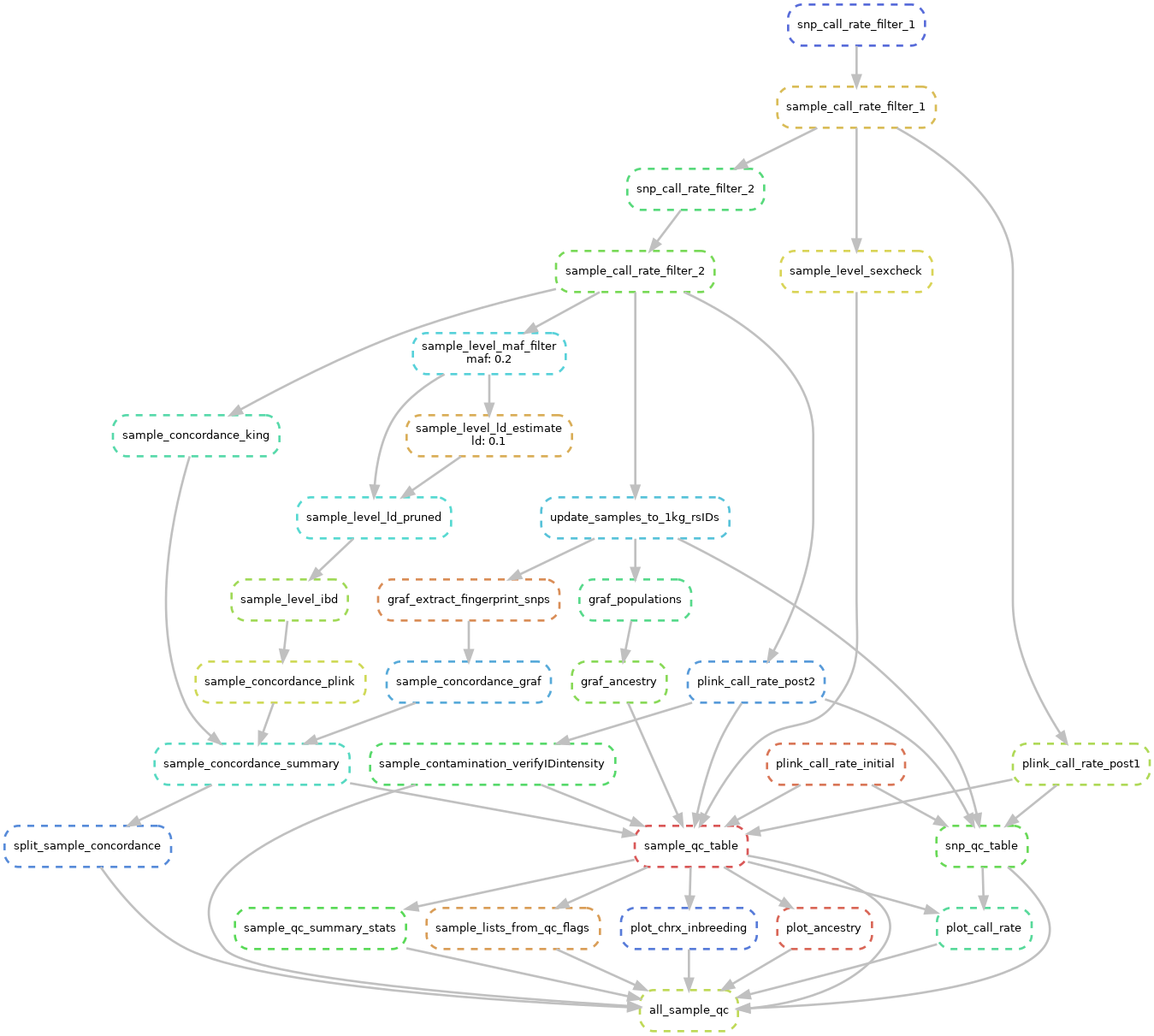
Fig. 4 The sample QC sub-workflow. This runs all major QC checks at the sample level.¶
Call Rate Filters¶
Config Options: see The config.yml for more details
software_params.sample_call_rate_1
software_params.snp_call_rate_1
software_params.sample_call_rate_2
software_params.snp_call_rate_2
Major Outputs:
sample_level/call_rate_1/samples.{bed,bim,fam}data set after level 1 sample/snp call rate filters.
sample_level/call_rate_2/samples.{bed,bim,fam}data set after level 2 sample/snp call rate filters.
First we run a series of call rate (a.k.a. completion) filters. Here we remove samples then SNPs which have high missingness in across samples.
Contamination Summary¶
Config Options: see The config.yml for more details
software_params.intensity_threshold
software_params.contam_threshold
Major Outputs:
sample_level/contamination/summary.csvaggregate summary table of contamination results.
If IDAT/GTC files are present and we ran Contamination Sub-workflow then we will create a summary table.
For samples that have a low call rate and a median idat intensity < the intensity_threshold we will set %Mix to NaN.
We then will flag a sample is_contaminated if the %Mix is >= contam_threshold.
Replicate Concordance¶
Config Options: see The config.yml for more details
software_params.ibd_pi_hat_min
software_params.ibd_pi_hat_max
software_params.maf_for_ibd
software_params.ld_prune_r2
software_params.dup_concordance_cutoff
software_params.pi_hat_threshold
workflow_params.remove_contam
workflow_params.remove_rep_discordant
workflow_params.concordance_tools
Major Outputs:
sample_level/concordance/summary.csv
It is common to include multiple replicates for a subset of samples as a QC metric.
Here we check these replicates and make sure that they are highly concordant.
The pipeline can check for relatedness/concordance using three tools : PLINK IBD, GRAF, and KING.
We only rely on and report IBD results in our summary tables.
To save compute resources on large datasets, the outputs for GRAF and KING relatedness checks can be disabled
by setting workflow_params.concordance_tools.graf and workflow_params.concordance_tools.king to False.
If PLINK IBD is not needed either, workflow_params.concordance_tools.plink can also be set to False.
In this case, PLINK IBD will be skipped. Since PLINK IBD is the main tool used for replicate checks,
columns related to replicate checks will be empty in the sample_qc table including Expected Replicate Discordance and Unexpected Replicate.
The workflow will successfully complete the sample_qc with all other QC metrics but not proceed to subject QC.
In the summary tables, we flag two samples as concordant (is_ge_concordance) if concordance (proportion IBD2) > dup_concordance_cutoff.
We flag two samples as related (is_ge_pi_hat) if PLINK’s PI_HAT is >= pi_hat_threshold.
Ancestry¶
Major Outputs:
sample_level/ancestry/graf_ancestry.txt
Finally, we estimate ancestry using GRAF. Here we categorize each sample into 1 of 9 possible groups (see GRAF for more details). We also report the percent mixture of the 3 major continental population (African, East Asian, European).
Sample QC Summary¶
Major Outputs:
sample_level/sample_qc.csv
sample_level/snp_qc.csv
sample_level/summary_stats.txt
sample_level/call_rate.png
sample_level/chrx_inbreeding.png
sample_level/ancestry.png
Finally, we aggregate all of the QC results. We generate two summary tables described below:
Internal Sample QC Report¶
Script: workflow/scripts/sample_qc_table.py
This script takes various outputs from the GwasQcPipeline and aggregates/summarizes results into an internal QC report. Here we use a functional approach to build up the QC report. Each file has it’s own function to parse/summarize its content. This makes it easier to add new components or change the behavior. Search for TO-ADD comments for where you would have to make modifications to add new components.
Output: sample_level/sample_qc.csv
name |
dtype |
description |
|---|---|---|
Sample_ID |
string |
The sample identifier used by the workflow. |
Group_By_Subject_ID |
string |
The subject identifier used by the workflow |
num_samples_per_subject |
UInt32 |
The total number of |
analytic_exclusion |
boolean |
True if the sample failed QC criteria. |
num_analytic_exclusion |
boolean |
The number of QC criteria the sample failed. |
analytic_exclusion_reason |
string |
A list of QC criteria the sample failed. |
is_subject_representative |
boolean |
True if the |
subject_dropped_from_study |
boolean |
True if there are no samples passing QC for this subject. |
case_control |
CASE_CONTROL_DTYPE |
Phenotype status [ |
is_internal_control |
boolean |
True if the sample is an internal control. |
is_sample_exclusion |
boolean |
True if the sample is excluded in the config or missing IDAT/GTC files. |
is_user_exclusion |
boolean |
True if the sample is excluded in the config |
is_missing_idats |
boolean |
True if the IDAT files were missing during pre-flight. |
is_missing_gtc |
boolean |
True if the IDAT files were missing during pre-flight. |
Call_Rate_Initial |
float |
The Initial sample call rate before any filters. |
Call_Rate_1 |
float |
The sample call rate after the first filter. |
Call_Rate_2 |
float |
The sample call rate after the second filter. |
is_cr1_filtered |
boolean |
True if a sample was removed by the first call rate filter. |
is_cr2_filtered |
boolean |
True if a sample was removed by the second call rate filter. |
is_call_rate_filtered |
boolean |
True if a sample was removed by either call rate filter. |
IdatIntensity |
float |
The median IDAT intensity. |
Contamination_Rate |
float |
The SNPweights contamination rate (%Mix) |
is_contaminated |
boolean |
True if a sample exceed the contamination rate threshold. |
replicate_ids |
string |
Concatenated Sample_IDs that are replicates. |
is_discordant_replicate |
boolean |
True if the replicates had low concordance. |
expected_sex |
SEX_DTYPE |
The expected sex from the provided sample sheet. |
predicted_sex |
SEX_DTYPE |
The predicted sex based on X chromosome heterozygosity. |
X_inbreeding_coefficient |
float |
The X chromosome F coefficient. |
is_sex_discordant |
boolean |
True if expected and predicted sex did not match. |
AFR |
float |
Proportion African ancestry. |
EUR |
float |
Proportion European ancestry. |
ASN |
float |
Proportion East Asian ancestry. |
Ancestry |
category |
Assigned ancestral population (GRAF). |
identifiler_needed |
boolean |
True if the lab needs to run Identifiler. |
identifiler_reason |
string |
The QC problem that needs checked with Identifiler. |
Internal SNP QC Report¶
Script: workflow/scripts/snp_qc_table.py
This script takes various outputs from the GwasQcPipeline and aggregates/summarizes results into an internal SNP level QC report.
Output: sample_level/snp_qc.csv
name |
dtype |
description |
|---|---|---|
array_snp_id |
string |
The SNP ID as found in the BPM file. |
thousand_genomes_snp_id |
string |
The rsID found in the 1000 Genomes VCF. |
chromosome |
string |
The chromosome that the SNP is on. |
snp_cr1 |
float |
The SNP call rate results after the first filter. |
snp_cr1_removed |
boolean |
True if the SNP was removed by the first call rate filter. |
snp_cr2 |
float |
The SNP call rate results after the second filter. |
snp_cr2_removed |
boolean |
True if the SNP was removed by the second call rate filter. |