Entry Points Sub-workflow¶
- Workflow File:
Config Options: see The config.yml for more details
user_files.gtc_pattern
user_files.idat_pattern
user_files.ped
user_files.map
user_files.bed
user_files.bim
user_files.fam
user_files.bcf
Major Outputs:
sample_level/samples.bed
sample_level/samples.bim
sample_level/samples.fam
The pipeline is an end-to-end workflow. It can accept raw IDAT files and generate a QC report. However, it can also continue from various other stages in analysis such as per-sample GTC files or an aggregated dataset file:
per-sample IDAT files:
Given user_files.idat_pattern and workflow_params.convert_idat2gtc=true, it would use the Illumina’s dragen array software to convert idats to gtcs and subsequently convert gtcs to aggregated BED/BIM/FAM.
To start with idat files, a cluster egt file using reference_files.illumina_cluster_file must be provided and dragen array sofware should be accesible either as module or a path provided using workflow_params.dragena_location
per-sample GTC files:
The pipeline supports two different methods for converting per-sample GTCs to aggregated BED/BIM/FAM:
If GTC files are provided using
user_files.gtc_patternandworkflow_params.convert_gtc2bcf=false(default) then following rulegraph will be followed:
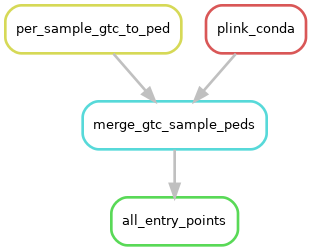
Fig. 2 The entry-point workflow with GTCs and convert_gtc2bcf=false.
If per sample GTC files are provided and convert_gtc2bcf is false, then we will convert these files to the PED/MAP format and merge them together.¶
If GTC files are provided using
user_files.gtc_patternandworkflow_params.convert_gtc2bcf=truethen the following rulegraph will be followed:

Fig. 3 The entry-point workflow with GTCs and convert_gtc2bcf=true.
If per sample GTC files are provided and convert_gtc2bcf is true, then we will convert these files to an aggregated BCF file and load the BCF file into Plink to create a BED/BIM/FAM set.¶
Aggregated dataset:
If a reanalysis for previous dataset is desired or GTC files are unavailable, an aggregated file encoding genotypes for all samples can be provided. The pipeline currently supports following three aggregated file formats:
If an aggregated PED/MAP is provided using
user_files.pedanduser_files.mapthen we will convert the PED/MAP to BED/BIM/FAM.If an aggregated BED/BIM/FAM is provided using
user_files.bed,user_files.bim,user_files.famthen we will create a symbolic link.If an aggregated BCF file is provided using
user_files.bcfthen we will convert the BCF to BED/BIM/FAM.